scSLAM-seq reveals core features of transcription dynamics in single cells
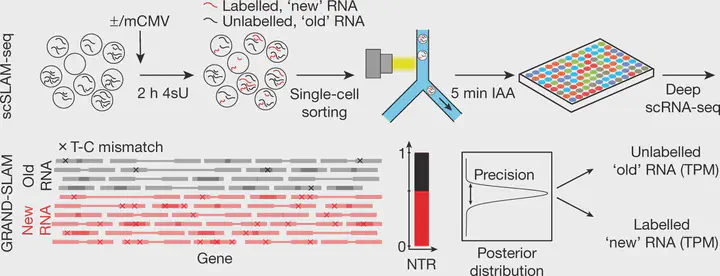 scSLAM-seq and GRAND-SLAM differentiate new from total RNA in single-cell studies. This provides an experimental alternative to RNA velocity.
scSLAM-seq and GRAND-SLAM differentiate new from total RNA in single-cell studies. This provides an experimental alternative to RNA velocity.Abstract
Single-cell RNA sequencing (scRNA-seq) has highlighted the important role of intercellular heterogeneity in phenotype variability in both health and disease1. However, current scRNA-seq approaches provide only a snapshot of gene expression and convey little information on the true temporal dynamics and stochastic nature of transcription. A further key limitation of scRNA-seq analysis is that the RNA profile of each individual cell can be analysed only once. Here we introduce single-cell, thiol-(SH)-linked alkylation of RNA for metabolic labelling sequencing (scSLAM-seq), which integrates metabolic RNA labelling2, biochemical nucleoside conversion3 and scRNA-seq to record transcriptional activity directly by differentiating between new and old RNA for thousands of genes per single cell. We use scSLAM-seq to study the onset of infection with lytic cytomegalovirus in single mouse fibroblasts. The cell-cycle state and dose of infection deduced from old RNA enable dose–response analysis based on new RNA. scSLAM-seq thereby both visualizes and explains differences in transcriptional activity at the single-cell level. Furthermore, it depicts ‘on–off’ switches and transcriptional burst kinetics in host gene expression with extensive gene-specific differences that correlate with promoter-intrinsic features (TBP–TATA-box interactions and DNA methylation). Thus, gene-specific, and not cell-specific, features explain the heterogeneity in transcriptomes between individual cells and the transcriptional response to perturbations.